| MitImpact id |
MI.15259 |
MI.15260 |
MI.15258 |
| Chr |
chrM |
chrM |
chrM |
| Start |
10197 |
10197 |
10197 |
| Ref |
G |
G |
G |
| Alt |
A |
C |
T |
| Gene symbol |
MT-ND3 |
MT-ND3 |
MT-ND3 |
| Extended annotation |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 3 |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 3 |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 3 |
| Gene position |
139 |
139 |
139 |
| Gene start |
10059 |
10059 |
10059 |
| Gene end |
10404 |
10404 |
10404 |
| Gene strand |
+ |
+ |
+ |
| Codon substitution |
GCC/ACC |
GCC/CCC |
GCC/TCC |
| AA position |
47 |
47 |
47 |
| AA ref |
A |
A |
A |
| AA alt |
T |
P |
S |
| Functional effect general |
missense |
missense |
missense |
| Functional effect detailed |
missense |
missense |
missense |
| OMIM id |
516002 |
516002 |
516002 |
| HGVS |
NC_012920.1:g.10197G>A |
NC_012920.1:g.10197G>C |
NC_012920.1:g.10197G>T |
| HGNC id |
7458 |
7458 |
7458 |
| Respiratory Chain complex |
I |
I |
I |
| Ensembl gene id |
ENSG00000198840 |
ENSG00000198840 |
ENSG00000198840 |
| Ensembl transcript id |
ENST00000361227 |
ENST00000361227 |
ENST00000361227 |
| Ensembl protein id |
ENSP00000355206 |
ENSP00000355206 |
ENSP00000355206 |
| Uniprot id |
P03897 |
P03897 |
P03897 |
| Uniprot name |
NU3M_HUMAN |
NU3M_HUMAN |
NU3M_HUMAN |
| Ncbi gene id |
4537 |
4537 |
4537 |
| Ncbi protein id |
YP_003024033.1 |
YP_003024033.1 |
YP_003024033.1 |
| PhyloP 100V |
4.687 |
4.687 |
4.687 |
| PhyloP 470Way |
0.848 |
0.848 |
0.848 |
| PhastCons 100V |
1 |
1 |
1 |
| PhastCons 470Way |
0.007 |
0.007 |
0.007 |
| PolyPhen2 |
probably_damaging |
probably_damaging |
probably_damaging |
| PolyPhen2 score |
1.0 |
1.0 |
1.0 |
| SIFT |
neutral |
neutral |
neutral |
| SIFT score |
0.52 |
0.32 |
1.0 |
| SIFT4G |
Damaging |
Damaging |
Damaging |
| SIFT4G score |
0.0 |
0.001 |
0.007 |
| VEST |
Neutral |
Pathogenic |
Neutral |
| VEST pvalue |
0.17 |
0.04 |
0.26 |
| VEST FDR |
0.45 |
0.35 |
0.45 |
| Mitoclass.1 |
damaging |
damaging |
neutral |
| SNPDryad |
Pathogenic |
Pathogenic |
Pathogenic |
| SNPDryad score |
0.93 |
0.96 |
0.98 |
| MutationTaster |
Disease automatic |
Polymorphism |
Polymorphism |
| MutationTaster score |
3.23265e-07 |
1 |
0.999992 |
| MutationTaster converted rankscore |
0.08975 |
0.08975 |
0.08975 |
| MutationTaster model |
complex_aae |
complex_aae |
complex_aae |
| MutationTaster AAE |
A47T |
A47P |
A47S |
| fathmm |
Tolerated |
Tolerated |
Tolerated |
| fathmm score |
0.89 |
0.82 |
0.98 |
| fathmm converted rankscore |
0.45636 |
0.48142 |
0.42122 |
| AlphaMissense |
likely_benign |
ambiguous |
likely_benign |
| AlphaMissense score |
0.2513 |
0.4019 |
0.1583 |
| CADD |
Deleterious |
Deleterious |
Deleterious |
| CADD score |
4.196092 |
3.865291 |
3.717795 |
| CADD phred |
23.9 |
23.5 |
23.3 |
| PROVEAN |
Damaging |
Damaging |
Damaging |
| PROVEAN score |
-3.52 |
-4.73 |
-2.76 |
| MutationAssessor |
medium |
medium |
low |
| MutationAssessor score |
2.045 |
2.03 |
1.14 |
| EFIN SP |
Damaging |
Damaging |
Damaging |
| EFIN SP score |
0.094 |
0.39 |
0.456 |
| EFIN HD |
Neutral |
Damaging |
Neutral |
| EFIN HD score |
0.38 |
0.236 |
0.366 |
| MLC |
Deleterious |
Deleterious |
Deleterious |
| MLC score |
0.55232663 |
0.55232663 |
0.55232663 |
| PANTHER score |
0.309 |
. |
. |
| PhD-SNP score |
0.797 |
. |
. |
| APOGEE1 |
Pathogenic |
Neutral |
Neutral |
| APOGEE1 score |
0.88 |
0.46 |
0.39 |
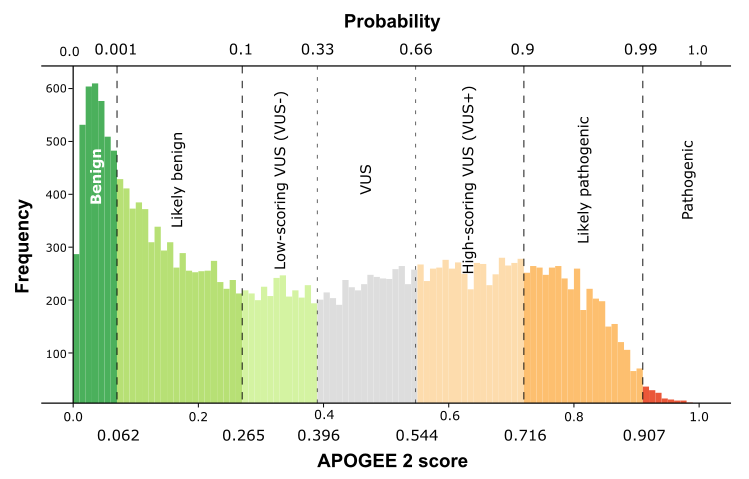
| APOGEE2 |
Likely-pathogenic |
VUS+ |
VUS |
| APOGEE2 score |
0.843686040876126 |
0.645176820055587 |
0.498126916021381 |
| CAROL |
deleterious |
deleterious |
deleterious |
| CAROL score |
1.0 |
1.0 |
1.0 |
| Condel |
neutral |
neutral |
deleterious |
| Condel score |
0.26 |
0.16 |
0.5 |
| COVEC WMV |
deleterious |
deleterious |
neutral |
| COVEC WMV score |
1 |
1 |
-2 |
| MtoolBox |
deleterious |
deleterious |
deleterious |
| MtoolBox DS |
0.79 |
0.84 |
0.75 |
| DEOGEN2 |
Damaging |
Damaging |
Tolerated |
| DEOGEN2 score |
0.52544 |
0.539619 |
0.495506 |
| DEOGEN2 converted rankscore |
0.83481 |
0.84213 |
0.81820 |
| Meta-SNP |
Disease |
. |
. |
| Meta-SNP score |
0.623 |
. |
. |
| PolyPhen2 transf |
low impact |
low impact |
low impact |
| PolyPhen2 transf score |
-3.43 |
-3.43 |
-3.43 |
| SIFT_transf |
medium impact |
medium impact |
high impact |
| SIFT transf score |
0.21 |
0.01 |
1.85 |
| MutationAssessor transf |
medium impact |
medium impact |
medium impact |
| MutationAssessor transf score |
1.22 |
1.39 |
0.51 |
| CHASM |
Neutral |
Neutral |
Neutral |
| CHASM pvalue |
0.76 |
0.31 |
0.21 |
| CHASM FDR |
0.85 |
0.8 |
0.8 |
| ClinVar id |
9715.0 |
. |
. |
| ClinVar Allele id |
24754.0 |
. |
. |
| ClinVar CLNDISDB |
MONDO:MONDO:0027068,MedGen:C4746992,OMIM:500014|MedGen:CN169374|MedGen:CN517202|MONDO:MONDO:0044970,MedGen:C0751651,Orphanet:68380|MedGen:CN043634|MONDO:MONDO:0009723,MedGen:C0023264,OMIM:256000,Orphanet:506|MONDO:MONDO:0010772,MedGen:C1839040,OMIM:500001,Orphanet:99718 |
. |
. |
| ClinVar CLNDN |
Mitochondrial_complex_1_deficiency,_mitochondrial_type_1|not_specified|not_provided|Mitochondrial_disease|Mitochondrial_DNA-Associated_Leigh_Syndrome_and_NARP|Leigh_syndrome|Leber_optic_atrophy_and_dystonia |
. |
. |
| ClinVar CLNSIG |
Pathogenic |
. |
. |
| MITOMAP Disease Clinical info |
Leigh Disease / Dystonia / Stroke / LDYT |
Leigh Disease |
. |
| MITOMAP Disease Status |
Cfrm [P] |
Reported |
. |
| MITOMAP Disease Hom/Het |
+/+ |
-/+ |
./. |
| MITOMAP General GenBank Freq |
0.0049% |
0.0% |
. |
| MITOMAP General GenBank Seqs |
3 |
0 |
. |
| MITOMAP General Curated refs |
21978175;37038312;30899856;28429146;30461153;30128709;17413873;18800376;17152068;18977334;35715829;30978515;15372108;19458970;29253894;11130070;30199507;32045392;38465286;20064630;12509511;21364701;30095618;30978516;20972245 |
38437941 |
. |
| MITOMAP Variant Class |
disease |
disease |
. |
| gnomAD 3.1 AN |
56422.0 |
56433.0 |
. |
| gnomAD 3.1 AC Homo |
0.0 |
0.0 |
. |
| gnomAD 3.1 AF Hom |
0.0 |
0.0 |
. |
| gnomAD 3.1 AC Het |
0.0 |
0.0 |
. |
| gnomAD 3.1 AF Het |
0.0 |
0.0 |
. |
| gnomAD 3.1 filter |
npg |
npg |
. |
| HelixMTdb AC Hom |
0.0 |
. |
. |
| HelixMTdb AF Hom |
0.0 |
. |
. |
| HelixMTdb AC Het |
7.0 |
. |
. |
| HelixMTdb AF Het |
3.5717385e-05 |
. |
. |
| HelixMTdb mean ARF |
0.15194 |
. |
. |
| HelixMTdb max ARF |
0.25 |
. |
. |
| ToMMo 54KJPN AC |
. |
. |
. |
| ToMMo 54KJPN AF |
. |
. |
. |
| ToMMo 54KJPN AN |
. |
. |
. |
| COSMIC 90 |
. |
. |
. |
| dbSNP 156 id |
rs267606891 |
. |
. |